How to download large number of single cell data
Many researchers submit the genomics data to sequence read archive (SRA). When the study involves sequencing of many single cells, downloading all of the files manually may be troublesome to do.
In this post, I will describe how to write a simple Python script to download a large number of files from European Nucleotide Archive.
Searching for the data
First, we need to search for the dataset using the accession number. I am interested in the dataset used by this paper; the accession number for the dataset is SRP114962.
Searching for the accession number at the ENA website leads to the following screen:
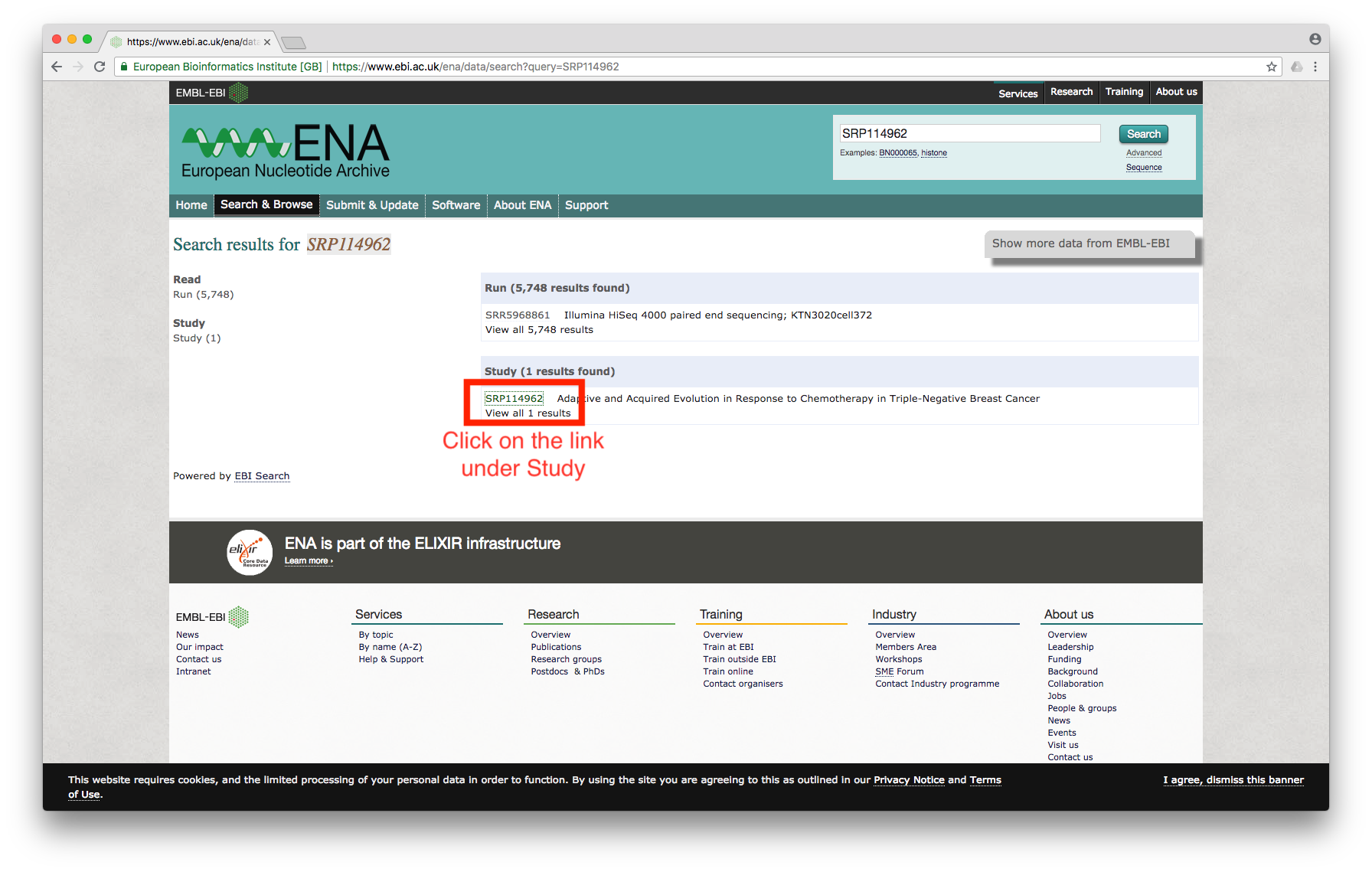 Figure 1
Figure 1
Click on the study, which will bring us to the page showing all of the files associated with the study:
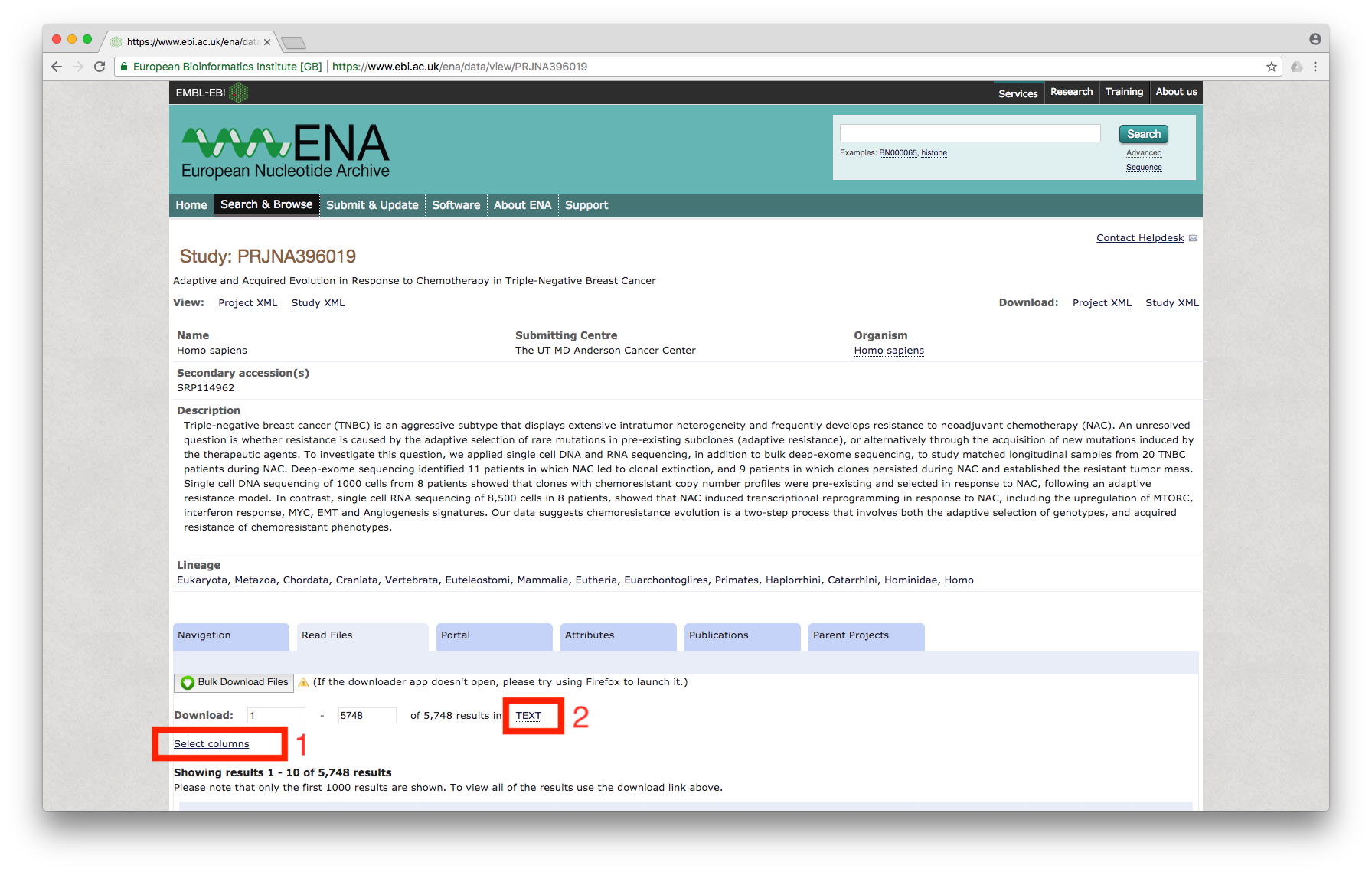 Figure 2
Figure 2
First, we will click on Select columns link (marked 1 in the above image) and enable the check box for Library name and Fastq bytes:
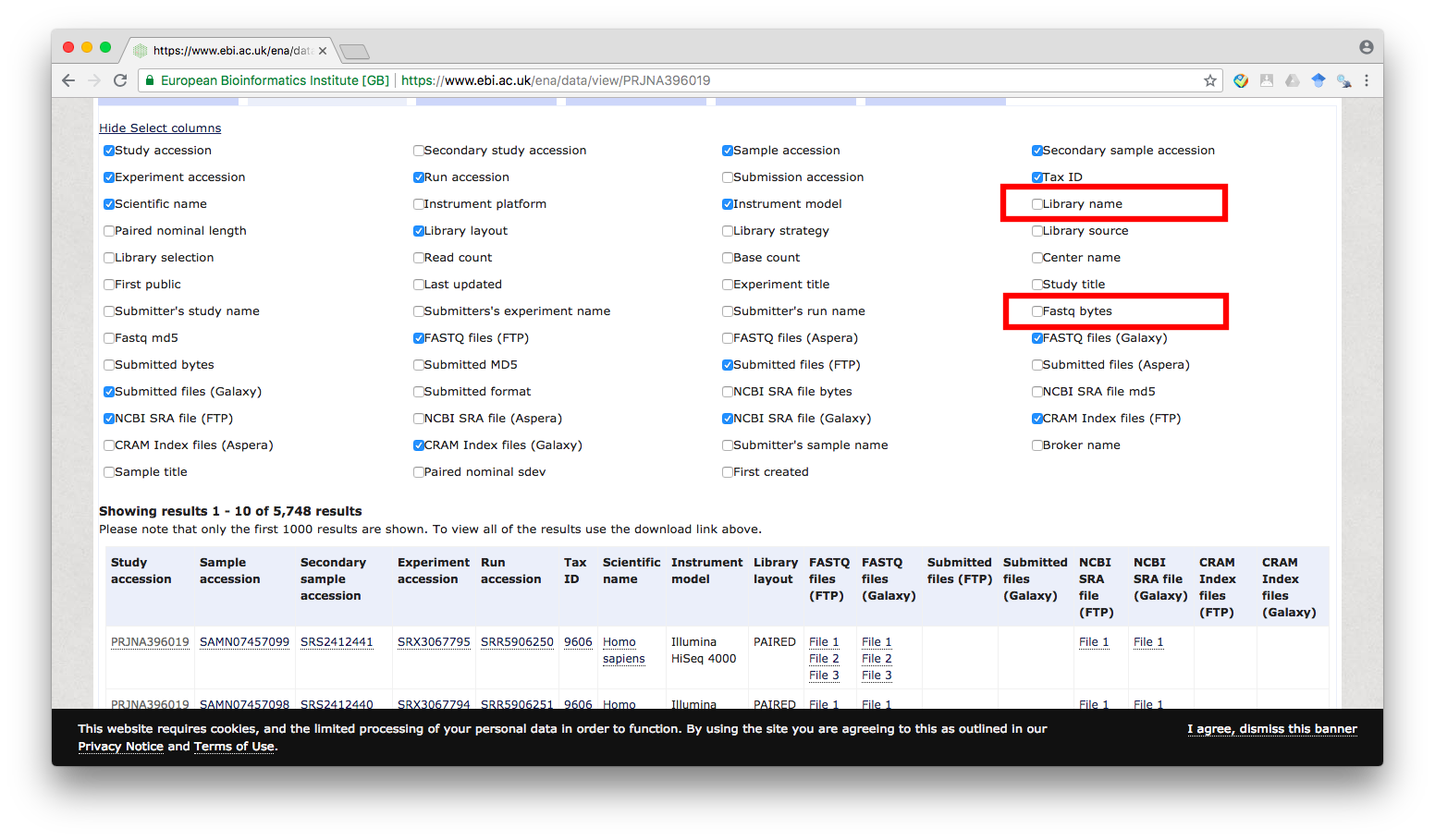 Figure 3
Figure 3
Then, click on TEXT (marked 2 in Figure 2), which will prompt for download. Download the file, name it as `table.txt’.
Open the downloaded file. It contains information associated with each file in the study. For example, FTP location for the file is under FASTQ files (FTP) column . The column Library name contains useful information for identifying the sample. For example, KTN102Blood identifies patient ID 102 and that the file corresponds to the Blood sample. The identifier used varies from dataset to dataset and the details are usually available in the paper or its supplement.
Note that you may unselect some of the other columns if they are unnecessary for you. In this post, only the Library name, FASTQ files (FTP), and Fastq bytes columns are required. For other dataset, you may need to select additional columns to help identify the files that you want to download.
Downloading the data
Most genomics data are large in size and hence, downloading all of the files in serial can take some time. Furthermore, processing of such genomics data usually takes place on a compute grid equipped with large amounts of memory and CPU cores. I will assume that the files are to be downloaded to a compute grid so that these files can be processed at a later time. Specifically, I will describe how to write a simple download script to be submitted to a grid sytem that uses job scheduler SLURM, as is the case with UPPMAX.
Step 1: Explore the master file
First, we need to write code to explore the master file to determine which files need to be downloaded. I have written this code using Jupyter Notebook and it is provided in a separate post.
Step 2: Preparing a SLURM script
There are two ways to submit a job. The first approach is to submit the command that you wish to execute directly using --wrap flag. For example:
sbatch -A project_id -p core -n 1 -t 1:00:00 -J download_ena -o log.out -e log.err --wrap "wget ftp_loc -O dest_file"
However, I think it may be better to write a separate script in case additional work is to be carried out on the downloaded file, for example, FastQC for quality checking. The simplest possible script for our purpose is as follows:
#!/bin/bash -l
echo "$(date) Running on: $(hostname)"
wget $1 -O $2
echo "$(date) File download complete."
We will pass in the source file location and destination as arguments to this script, which will be generated from Python.
Step 3: Writing Python script to generate job submission commands
We will write a script to read in the information in table.txt and generate job submission commands for SLURM. First, we will import the necessary libraries.
import numpy as np
import pandas as pd
import argparse
import os
import subprocess
import time
import sys
We will require the destination directory, location of the master file, patient ID to be downloaded, SLURM project ID to be provided as command line arguments. Additionally, we can require additional information such as job name, number of hours required for the job, and whether to launch the job or generate string commands for debug purposes. We will use argparse library to simplify processing of the command line arguments:
parser = argparse.ArgumentParser(description='Process file destination.')
parser.add_argument('dest', help="Specify the destination directory.", type=str)
parser.add_argument('master_file', help="Specify the master file downloaded from ENA.", type=str)
parser.add_argument('patient_id', help="Specify the patient ID. E.g. KTN102.", type=str)
parser.add_argument('project_id', help="Project ID.", type=str)
parser.add_argument('--debug', help="Enter yes/no. If no is provided, it will only print the command", type=str)
parser.add_argument('--time', help="Number of hours per job.", type=str, default="4:00:00")
parser.add_argument('--job_name', help="Job name.", type=str, default="wget_sc")
args = parser.parse_args()
We will now load the master file using pandas, and extract out only the records corresponding to the patient ID:
# read in the master file and extract the files that we want
df = pd.read_table(args.master_file, sep='\t')
patient_ids = df['library_name'].astype(str).str[0:6]
# extract records corresponding to the specified patient
df2 = df.loc[patient_ids == args.patient_id]
Let’s create new directories for better organization:
# make a directory if it does not exist alrady
if not os.path.exists(args.dest):
os.mkdir(args.dest)
# make a directory for patient_id if it does not exist already
download_dest = os.path.join(args.dest, args.patient_id)
print(download_dest)
if not os.path.exists(download_dest):
os.mkdir(download_dest)
# generate a directory to hold just the scRNA data files to be downloaded
rna_download_dest = os.path.join(download_dest, "scRNA")
if not os.path.exists(rna_download_dest):
os.mkdir(rna_download_dest)
# generate the log files for each download job
logs = os.path.join(rna_download_dest, "logs")
if not os.path.exists(logs):
os.mkdir(logs)
Finally, generate the SLURM commands and submit the job (unless we are in a debug mode):
# code to download all RNA data files
for idx, row in df2.iterrows():
fastq_ftp_loc = row['fastq_ftp']
locs = fastq_ftp_loc.split(";")
library_name = row['library_name']
if len(locs) == 1: # scRNA data are in one fastq.gz file
accession_number = row["run_accession"]
# retrieve the file name and form the destination path
loc_split = locs[0].split("/")
dest_file_name = loc_split[len(loc_split)-1]
final_dest = os.path.join(rna_download_dest, dest_file_name)
command = "sbatch -A %s -p core -n 1 -t %s -J %s -o %s/%s.out -e %s/%s.err worker.sbatch %s %s" % (args.project_id, args.time, args.job_name, logs, accession_number, logs, accession_number, locs[0], final_dest)
if (args.debug is None or args.debug == "no"):
print(command)
else:
# submit the job
subprocess.call([command], shell=True)
To explain, we noted in this post that scRNA files are stored as a single file whereas scDNA and bulk DNA files are organized into 3 files. Therefore, if a record is associated with only one file, we download the file. Note that this approach requires carefully checking over the submission file patterns in advance and the code will have to be changed for different dataset.
The entire Python code is available here and the SLURM script is available here.